Perbezaan antara talasemia alfa dan beta
Perbezaan Utama - Alpha Vs Beta Thalassemia
Thalassemia adalah kumpulan gangguan heterogen yang disebabkan oleh mutasi yang diwarisi yang mengurangkan sintesis sama ada rantai globin alpha atau beta, yang membawa kepada anemia, hipoksia tisu dan hemolisis sel merah yang berkaitan dengan ketidakseimbangan dalam sintesis rantai globin. Terdapat dua bentuk utama talasemia sebagai thalassemia alpha dan thalassemia beta. Dalam talasemia alfa, terdapat penurunan bilangan rantai globin alfa sedangkan dalam beta-talasemia ia adalah bilangan rantai beta globin yang turun. Ini adalah perbezaan utama antara alpha dan beta talasemia.
Kandungan
1. Gambaran Keseluruhan dan Perbezaan Utama
2. Apa alpha thalassemia
3. Apa Beta Thalassemia
4. Persamaan antara alpha dan beta talasemia
5. Perbandingan sampingan - Alpha vs beta thalassemia dalam bentuk jadual
6. Ringkasan
Apa itu alpha thalassemia?
Di Alpha Thalassemia, beberapa gen yang bertanggungjawab untuk pengekodan rantai globin alfa. Gen Globin Alpha umumnya mempunyai empat salinan. Keparahan penyakit bergantung pada jumlah salinan yang hilang.
Hydrops fetalis
Sintesis rantai globin alfa ditindas sepenuhnya apabila semua empat salinan gen globin alpha hilang. Oleh kerana rantai globin alfa diperlukan untuk sintesis kedua -dua hemoglobin janin dan dewasa, keadaan ini tidak serasi dengan kehidupan; oleh itu dalam penamatan utero kehamilan berlaku jika janin dipengaruhi oleh keadaan ini.
Penyakit HBH
Keadaan ini disebabkan oleh ketiadaan tiga salinan gen globin alpha. Ini mengakibatkan anemia microcytic hypochromic yang sederhana dan teruk dengan splenomegali yang berkaitan.
Ciri -ciri Alpha Thalassemia
Ini disebabkan oleh ketiadaan atau tidak aktif satu atau dua salinan gen globin alpha. Walaupun sifat talasemia alfa tidak menyebabkan anemia, mereka dapat mengurangkan jumlah volum korpuskular dan tahap hemoglobin korpuskular sambil meningkatkan kiraan sel darah merah melebihi 5.5*1012/L.
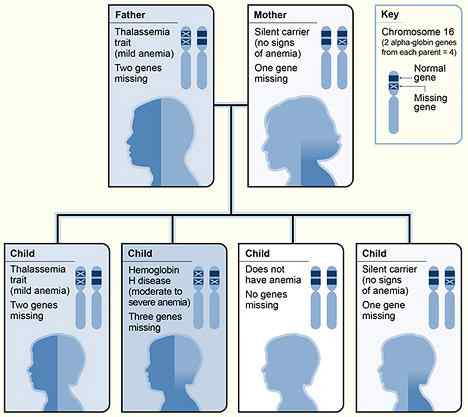
Rajah 01: Warisan alpha thalassemia
Diagnosis Alpha Thalassemia adalah melalui kajian sintesis rantai globin.
Pengurusan
Pesakit dengan bentuk anemia ringan biasanya tidak memerlukan sebarang rawatan. Pentadbiran Besi dan Asid Folik hanya dianjurkan pada sesetengah pesakit. Mereka yang mempunyai bentuk alpha thalassemia yang teruk memerlukan pemindahan darah seumur hidup.
Apa itu thalassemia beta?
Dalam talasemia beta, jumlah rantai globin beta turun.
Beta Thalassemia Major
Jika kedua -dua ibu bapa adalah pembawa sifat talasemia beta, kemungkinan anak talasemia beta yang mewarisi adalah 25%. Di Beta Thalassemia Major, pengeluaran rantai Globin beta sama ada ditindas sepenuhnya atau dikurangkan secara drastik. Oleh kerana tidak ada rantai globin beta yang cukup untuk mereka bergabung, rantai globin alpha yang berlebihan dapat disimpan dalam sel -sel merah yang matang dan tidak matang. Ini membawa kepada hemolisis pramatang sel merah dan erythropoiesis yang tidak berkesan.
Ciri -ciri klinikal
- Anemia teruk, yang menjadi jelas pada 3-6 bulan selepas kelahiran.
- Splenomegaly dan Hepatomegaly
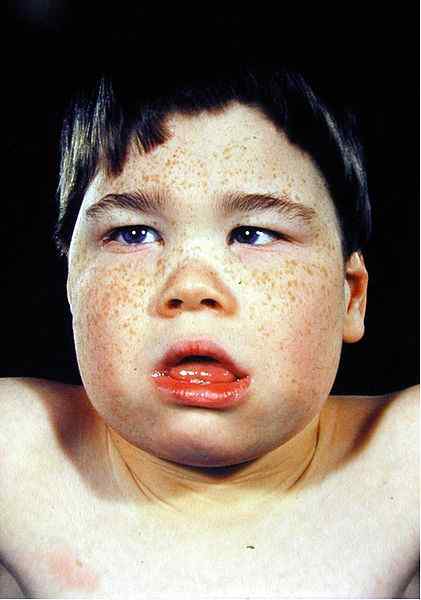
- Facies thalassemic
Perubahan dalam ciri -ciri wajah adalah disebabkan oleh pengembangan tulang kerana hiperplasia sumsum tulang. Radiograf X-ray menunjukkan penampilan rambut tengkorak yang biasanya dilihat dalam talasemia beta.
Rajah 02: Facies thalassemic
Diagnosis makmal
Kromatografi cecair berprestasi tinggi (HPLC) adalah kaedah utama yang digunakan dalam diagnosis penyakit hematologi pada masa kini. HPLC utama beta thalassemia menunjukkan kehadiran tahap HBA yang dikurangkan dengan tahap HBF yang luar biasa tinggi. Kiraan darah penuh akan mendedahkan kewujudan anemia microcytic hypochromic, dan pemeriksaan filem darah akan menunjukkan kehadiran peningkatan jumlah reticulocytes bersama -sama dengan stippling basofilik dan sel sasaran.
Rawatan
- Transfusi darah biasa
- Terapi Chelation Besi
- Asid folik (jika pengambilan asid folik diet tidak memuaskan)
- Splenectomy (kadang -kadang digunakan untuk mengurangkan keperluan darah)
- Transplantasi sumsum tulang
- Terapi gen untuk tujuan pemeriksaan dan terapeutik
Beta Thalassemia Ciri/Kecil
Beta thalassemia minor adalah keadaan biasa yang sering tidak gejala. Walaupun tanda -tanda dan gejala serupa dengan talasemia alfa, beta thalassemia lebih teruk daripada rakan sejawatannya. Diagnosis beta thalassemia minor dibuat jika HBA2 tahap lebih daripada 3.5%.
Thalassemia Intermedia
Intermedia thalassemia merujuk kepada kes -kes talasemia keterukan sederhana yang tidak memerlukan pemindahan biasa.
Apakah persamaan antara alpha dan beta talasemia?
- Dalam kedua -dua keadaan, terdapat penurunan tahap darah hemoglobin.
Apakah perbezaan antara alpha dan beta talasemia?
Alpha vs beta thalassemia | |
| Terdapat penurunan bilangan rantai globin alfa. | Terdapat penurunan bilangan rantaian globin beta. |
| Penghapusan gen | |
| Beberapa gen yang bertanggungjawab untuk pengekodan rantai globin alfa dipadamkan. | Gen yang bertanggungjawab untuk sintesis rantai globin beta sama ada sebahagiannya atau sepenuhnya dipadam. |
| Jenis | |
| Hydrops fetalis, penyakit HBH dan sifat alpha talasemia adalah bentuk utama alpha thalassemia. | Terdapat dua bentuk utama thalassemia beta sebagai beta thalassemia utama dan beta thalassemia minor. |
| Diagnosis | |
| Diagnosis Alpha Thalassemia adalah melalui kajian sintesis rantai globin. | Kromatografi cecair berprestasi tinggi (HPLC) adalah siasatan yang digunakan untuk diagnosis talasemia beta. |
| Ciri -ciri klinikal | |
|
|
| Rawatan dan Pengurusan | |
|
|
Ringkasan -Alpha vs Beta Thalassemia
Thalassemia adalah kumpulan gangguan heterogen yang disebabkan oleh mutasi yang diwarisi yang mengurangkan sintesis sama ada rantai globin alpha atau beta yang mengarang hemoglobin dewasa HBA. Thalassemia boleh dikategorikan secara meluas ke dalam dua kategori utama sebagai alpha talasemia dan beta talasemia. Dalam talasemia alfa, jumlah rantai alpha berkurangan, dan dalam beta-talasemia, bilangan rantai beta berkurangan. Ini adalah perbezaan utama antara alpha dan beta talasemia.
Muat turun versi pdf alpha vs beta thalassemia
Anda boleh memuat turun versi PDF artikel ini dan menggunakannya untuk tujuan luar talian mengikut nota petikan. Sila muat turun versi pdf di sini perbezaan antara alpha dan beta talasemia
Rujukan:
1.Kumar, Parveen J., dan Michael l. Clark. Perubatan Klinikal Kumar & Clark. Edinburgh: w.B. Saunders, 2009.
Ihsan gambar:
1. "Thalassemia Alpha" oleh National Heart Lung and Blood Institute (NIH) - National Heart Lung and Blood Institute (NIH) (Domain Awam) melalui Commons Wikimedia
2. "ATR-X" oleh Gibbons R. - Gibbons r. Alpha Thalassaemia-Mental Retardation, x dikaitkan. Orphanet J Rare Dis. 1, 15. 2006. doi: 10.1186/1750-1172-1-15. PMID 16722615 (CC oleh 2.0) melalui Commons Wikimedia